

新基淋巴瘤CAR-T疗法有望明年获批?!

美国时间11月7日,根据在2019年美国血液学会(ASH)官网上发布的摘要,新基公布了旗下CAR-T疗法一项关键试验的数据。这些数据看起来有望与吉利德/Kite的Yescarta和诺华的Kyrimah一争高下,由此分析师预测新基的CAR-T疗法有望在明年获得FDA的批准。
在这项I期关键试验中,262例大B细胞淋巴瘤患者接受抗CD19CAR-T疗法lisocabtagene maraleucel (liso-cel)。在255例可评价疗效的患者中,完全缓解率(CR)为53%,总缓解率(ORR)为73%,中位缓解时间为13.3个月。
liso-cel的缓解率比Kymriah说明书上给出的缓解率要高出20个百分点,比Yescarta的结果高出1-2个百分点。由于这三种药物是在不同的试验中研究的,因此以上交叉试验对比可能存在偏移。
投资银行Jefferies的分析师认为,liso-cel的数据足够强劲,足以将该药物推向市场。
“总之,liso-cel的试验结果看起来符合我们对疗效和安全性的预期,我们依然认为,成功获得批准的概率非常高。”这些分析师在给投资者的一份报告中写道。
liso-cel(以前称为JCAR017)的安全性数据可能会进一步增加其获批的可能性。虽然有4例死亡与liso-cel相关,但总体而言,治疗引起的不良事件发生率低于Kymirah和Yescarta试验。
在研发过程中,Kite称Yescarta 与87%的神经毒性率和13%的3级或更高的细胞因子释放综合征(CRS)有关。Liso-cel在试验中的神经毒性率和3级CRS分别为30%和2%。新基对Liso-cel的安全性和耐受性有足够的信心。
对CAR-T风险机制的进一步理解,可能是liso-cel和Yescarta之间安全性数据差异的原因——Yescarta是在细胞疗法的理解还处于非常早期的阶段时发展起来的。
不过新基在制备liso-cel的能力上似乎遇到不少挑战,可能会对liso-cel的审批产生负面影响。从分离白细胞到获得liso-cel的平均时间为24天,导致新基无法为两名患者及时制备liso-cel,并向24名患者提供不符合质量标准的产品。
这对新基产生不少压力。根据百时美施贵宝的收购条款,新基的liso-cel需要在2020年底获得批准,才能启动每股9美元的现金支付。
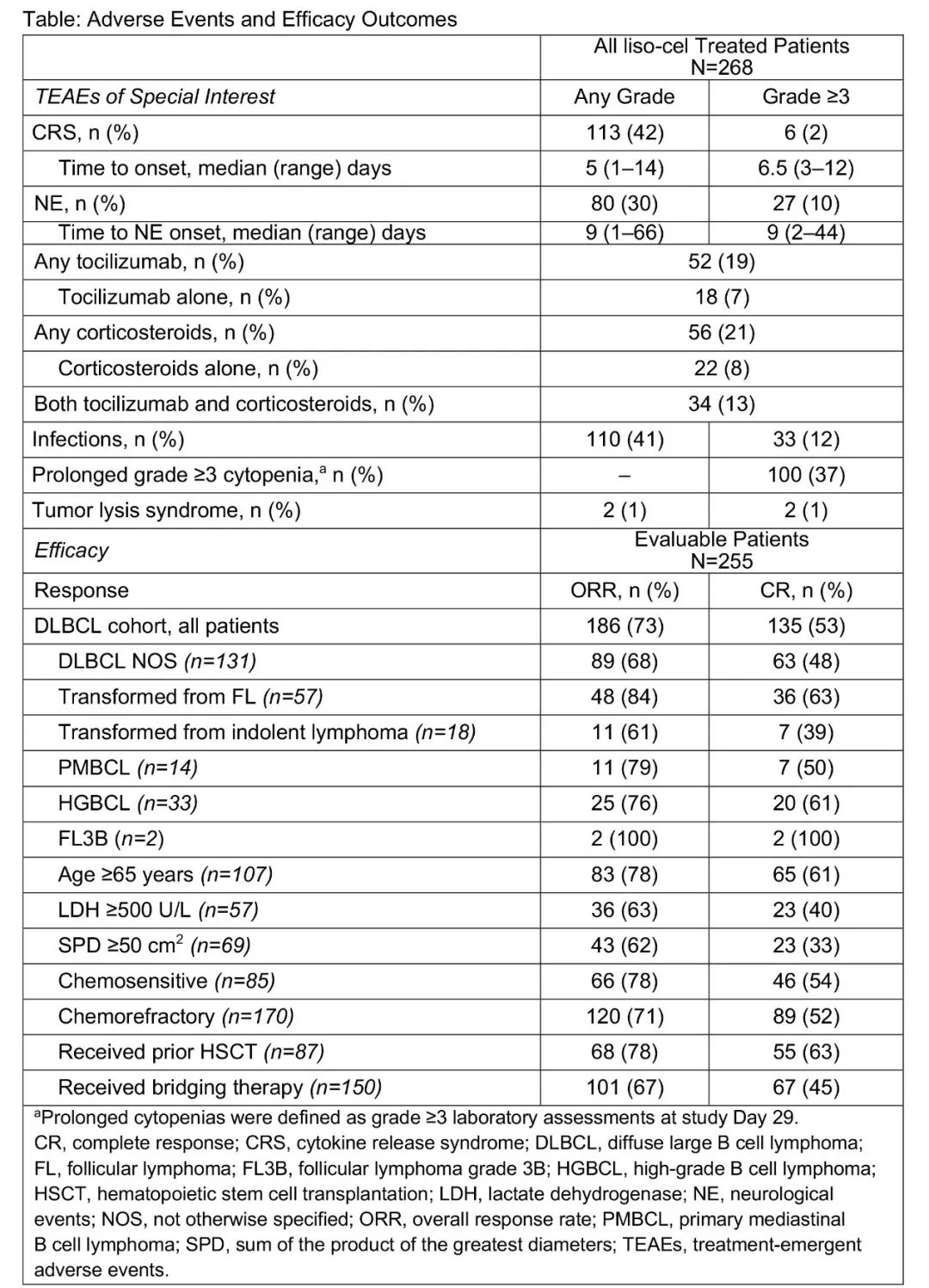
▲ 新 基CAR-T疗法liso-ce l对淋巴 瘤的疗效与安全性数据
a长期细胞减少被定义为研究日的实验室评估等级≥3级
CR:完全缓解;CRS:细胞因子释放综合征;DLBCL:弥漫性大B细胞淋巴瘤;
FL:滤泡性淋巴瘤;FL3B:滤泡性淋巴瘤3B级;
HGBCL:高级B细胞淋巴瘤;HSCT:造血干细胞移植
LDH:乳酸脱氢酶; NE:神经系统不良事件;
NOS:未另行说明; ORR:总缓解率; PMBCL:原发性纵隔B细胞淋巴瘤
SPD:最大直径乘积之和;TEAES:出现治疗不良反应事件
宾大首个基因编辑T细胞疗法初步人体试验安全性良好!

2016年,Facebook和Napster亿万富翁肖恩·帕克(Sean Parker)因承诺“击溃癌症”并给生命科学研究带来“颠覆性突破”而备受关注,但这不仅仅是空谈:这位互联网先驱斥资2.5亿美元成立了帕克癌症免疫治疗研究所(Parker Institute forCancer Immunology therapy),该研究所随后率先得到美国国立卫生研究所(NIH)批准——获批启动了一项利用基因编辑技术CRISPR-Cas9进行T细胞免疫肿瘤学疗法(NYCE T细胞)的人体试验。
即将在奥兰多举行的2019年美国血液学学会(ASH)年会上(2019年12月7-10日),由宾夕法尼亚大学艾布拉姆森癌症中心领导的研究小组,将分享参加该试验的两名多发性骨髓瘤患者和一名肉瘤患者的情况,该试验也得到了Tmunity Therapeutics公司的支持。
根据周三在ASH公布的一份研究摘要,受试者对该CRISPR-T细胞免疫疗法耐受性良好。在该试验中,基因编辑过的免疫细胞在增殖后注入到目标肿瘤中。而这三名都曾经历过治疗失败的受试者,在他们输注细胞疗法的九个月后仍然活着,尽管现在评估他们的整体响应还为时过早。
该试验的核心技术是利用CRISPR对从病人身上采集的T细胞进行三次编辑:
· 其中一项编辑消除了T细胞的PD-1——PD-1是一种可阻止免疫细胞识别和攻击癌细胞的“检查点”
· 另外两项剪去了T细胞的一部分,而剪去的部分正好有可能阻止T细胞附着在癌细胞上。
· 然后利用病毒插入一个“亲和增强型T细胞受体(TCR)(TCR可使T细胞定位癌细胞上的特定抗原)
宾夕法尼亚大学是CAR-T技术的先驱,正是在那里孕育出了诺华的CAR-T疗法Kymriah以及后来的商业化。但这次试验中使用的细胞不太一样。
Kymriah的CAR-T细胞嵌合了针对CD19抗原的受体,这使得这些改造后的CAR-T细胞能够攻击CD19抗原阳性的癌细胞。
相比之下,此次的CRISPR编辑过的T细胞只在HLA-A201阳性的情况下才有活性(HLA-A201是一种在某些癌症患者中会过度表达抗原)。因此,患者在接受治疗前必须进行HLA-A201表达状态检测。
帕克研究所资助了许多学术机构和公司的免疫肿瘤学研究。该组织最近的其他细胞疗法进展包括与Xyphos Biosciences达成协议,开发下一代CAR-T疗法;并参与了一笔8500万美元对ArsenalBio的资助,同样计划利用CRISPR开发细胞疗法。
此次在宾夕法尼亚大学的试验涉及18例多发性骨髓瘤、滑膜肉瘤或黏液样/圆形细胞脂肪肉瘤患者,预计将持续到2022年。研究人员计划在12月7日的ASH会议上提供更多关于细胞功能和临床结果的细节。
渤健和三星扩大生物仿制药交易范围,覆盖Lucentis、Eylea和中国市场

在与其仿制药合资企业三星Bioepis建立了成功的合作伙伴关系后,渤健决定将业务扩展到更多的药品和中国市场。
11月6日,渤健宣布与三星Bioepis达成一项新交易,获得后者两种新型眼科生物仿制药在美国、加拿大、欧洲、日本和澳大利亚等全球主要市场商业化的独家权利,这两款药物分别为SB11(罗氏和诺华的Lucentis®的生物仿制药)和SB15(再生元和拜耳的Eylea®的生物仿制药)。
根据交易条款,渤健将向三星Bioepis支付1亿美元的预付款,并可能会再支付2.1亿美元的额外开发、监管和基于销售的里程碑付款。三星Bioepis将负责开发,并将以预定的毛利率向渤健提供这两种产品。目前,Lucentis的候选生物仿制药正处于III期临床试验阶段, Eylea的候选生物仿制药仍处于临床前阶段。
此外,这次交易渤健将获得三星Bioepis的抗-TNF生物仿制药产品组合——Benepalitm(依那西普)、Flixabitm(英夫利昔单抗)和Imralditm(阿达木单抗)在中国的独家商业化权利,以换取该市场销售的特许权使用费;并且还将获得一项选择权——将这三个抗-TNF生物仿制药的当前欧洲商业协议期限再延长五年,但需支付6000万美元的选择权执行费。
渤健首席执行官Michel Vounatsos在一份声明中说:“我们对将生物仿制药引入一个新的治疗领域以及世界各地新区域的潜力感到兴奋,其目标是可持续地促进这些药物对有需要的患者的可及性”。
目前,渤健获得的抗-TNF产品组合只在欧盟上市,而且在那里的销售似乎遇到了瓶颈。第三季度,这三个抗-TNF生物仿制药共赚得1.84亿美元,与上季度持平,比分析师预期低7.5%。这主要是因为Enbrel(依那西普)的生物仿制药Benepalitm同比下降了6%,仅达到1.16亿美元。
这些治疗老年性黄斑变性和其他视力损害的眼部药物,与中国市场一起,可能会给渤健的生物仿制药特许经营权带来销量的提升。前9个月,罗氏和诺华的Lucentis的销售总额增至约29.4亿美元。至于Eylea,再生元在美国的销售额增长了12%,达到34.2亿美元,拜耳在世界其他地区的销售额增长了15%,达到18.3亿欧元(20.2亿美元)。
在中国,渤健可能需要加快一点速度。本周四,中国国家药品监督管理局(NMPD)通过了中国首家由本土药企百奥泰生产的Humira生物仿制药格乐立,以治疗银屑病、类风湿关节炎和强直性脊柱炎。百奥泰尚未取得任何营收,目前正申请科创板上市。
中国可能只占Humira 200亿美元年销售额的一小部分。Humira 于2010年在中国获得批准,Humira尚未进入国家医保目录。根据百奥泰的招股说明书,2018年原研药Humira在中国市场的平均价格为每单位40毫克/0.8毫升7586元人民币(1087美元),相当于每年约20万元人民币(28660美元)的治疗费用。
“高昂的价格限制了Humira的可及性,”该公司表示。
该公司指出,到今年5月,约有15家中国药企携其Humira生物仿制药进入临床试验。已提交上市申请的直接竞争对手包括信达生物和复宏汉霖——今年2月复宏汉霖的汉利康成为在中国首个获批的罗氏美罗华(利妥昔单抗)生物仿制药。
援引国际权威分析机构沙利文(Frost &Sullivan)的报告,百奥泰估计,到2023年,中国的Humira生物仿制药市场可能达到47亿元人民币(合6.74亿美元)。