
安乃近注射液有关物质检查
来源
中国药师 2018 年第21 卷第3期
作者
兰文,刘轶,黄靖
湖南省药品检验研究院
厦门市食品药品质量检验研究院
摘要
目的:
采用HPLC 法测定安乃近注射液的有关物质。
方法:
采用Xtimate C18( 250 mm × 4.6 mm,5 μm) 色谱柱,以磷酸盐缓冲液( 取磷酸二氢钠6.0 g,加水1 000 ml,加三乙胺1 ml,用氢氧化钠溶液调pH 至7.0) -甲醇( 75:25) 为流动相,流速为1.0 ml·min-1 ; 检测波长254 nm,柱温为30℃; 进样量为5 μl。
结果:
在该色谱条件下,空白辅料、杂质峰与主峰均能有效分离; 本品共检出2 个已知杂质( 杂质C 及杂质E) 及4 个未知杂质;
杂质C 的量在2.87% ~5.82% 之间,杂质E 的量均小于0.1%,4 个未知杂质量均小于0.1%。
结论:
本法简便、准确,专属性强,有助于更好地控制本品的质量。
关键词
安乃近注射液; 高效液相色谱法; 有关物质
正文 |
安乃近注射液为吡唑酮类解热止痛药,用于高热时的解热,也可用于头痛、偏头疼、肌肉痛、关节痛和痛经等[1,2]。
其主要成份为安乃近,化学名称为[( 1,5-二甲基-2-苯基-3-氧代-2,2-二氢-1-H-吡啶-4-基) 甲氨基] 甲烷磺酸钠盐一水合物(C13H16N3NaO4S·H2O) ,辅料为亚硫酸盐、依地酸二钠、苯甲醇和注射用水等。
安乃近注射液现行标准收载于中国药典2005 年版增补本[3],国外药典均未收载该品种。现行标准中未控制该品种的有关物质,作为不良反应较大的高风险注射剂,有必要对有关物质开展研究。
本文参考相关文献[4 ~10],建立了HPLC 法测定该品种的有关物质,并对检出的已知杂质的来源及毒性进行分析。现报道如下。
1
仪器与试药
1.1 仪器
Agilent1260 高效液相色谱仪,包括QUAT pump VL 泵系统、ALS 进样系统、TCC 柱温箱及VWD 检测器( 美国安捷伦科技有限公司) ; Dionex Ultimate3000 高效液相色谱仪,包括Ultimate3000 pump 系统、Autosampler 系统、RS Column Compartment及Diode Array Detector ( 美国戴安公司) ; ABS-265型电子分析天平( 瑞士梅特勒-托利多公司,分度值0.01 mg) 。
1.2 试药
安乃近对照品( 批号: 100002-201607,按C13H16N3NaO4 S计,供HPLC 法测定,含量为94.5%) 、4-甲氨基安替比林( 杂质C) 对照品( 批号: 101115-201101,按C12H15N3O 计,含量为96.6%) 、氨基比林( 杂质D) 对照品( 批号: 100503-201302,按C13H17O 计,含量为99.9%) 、4-N-去甲基安乃近( 杂质E)对照品( 批号: 100054-201104) 均由中国食品药品检定研究院提供;
4-甲酰氨基安替比林( 杂质A) 对照品( EP,批号:4.0) 、4-氨基安替比林( 杂质B) 对照品( TRC,批号: 1-LWJ-76-1) 、吡唑酮( 杂质F) 对照品( EP,批号: 1.0) 和安替比林( 杂质G) 对照品( 批号: 30104) 由Dr.Ehrenstorfer GmbH 提供; 甲醇( 色谱纯,Burdick & JacksonTM,批号: Q6CG1H) ;
其余试剂均为分析纯; 水为纯化水。安乃近注射液15 家生产企业各1 ~3 批样品,规格: 2 ml:0.5 g。
2
方法与结果
2.1 色谱条件
色谱柱: Xtimate C18柱( 250 mm × 4.6 mm,5 μm) ; 流动相: 磷酸盐缓冲液( 磷酸二氢钠6.0 g,加水1 000 ml,加三乙胺1 ml,用氢氧化钠溶液调pH 至7.0) -甲醇( 75:25) ; 检测波长: 254 nm; 流速: 1.0 ml·min-1 ; 柱温: 30℃; 进样量: 5μl。
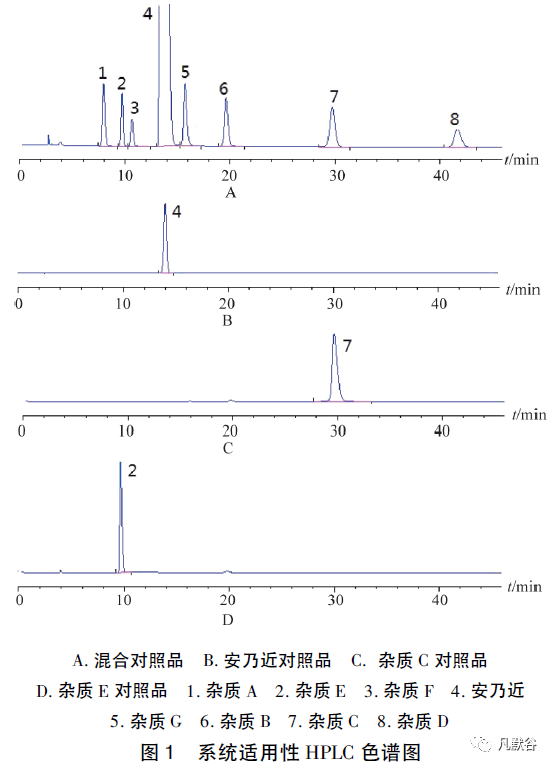
2.2 系统适用性试验
取安乃近对照品及各杂质对照品适量,用无水甲醇溶解并稀释制成10 μg·ml-1 的溶液,精密量取5 μl,按“2.1”项下色谱条件测定,色谱图见图1。

结果显示,在该色谱条件下安乃近与其主要杂质均能有效分离。
2.3 强制破坏试验
取样品( N 公司,批号: 16060672) 1.0 ml,分别置50 ml量瓶中,用强光( 于照度4 500 Lx ± 500 Lx 的条件下放置5d) 、强酸( 1 mol·L-1 盐酸溶液2 ml,室温放置1 h,并时时振摇,加1 mol·L-1 氢氧化钠溶液调至中性) 、强碱( 1 mol·L-1 氢氧化钠溶液2 ml,室温放置1 h,并时时振摇,加1 mol·L-1盐酸溶液调至中性) 、氧化( 3% 过氧化氢溶液1 ml,室温放置10 min,并时时振摇) 、高温( 置70 ~80℃水浴中放置1 h,放冷) 进行破坏。
结果表明: 本品对酸、碱、氧化、热、光均不稳定,在酸、碱、氧化及热破坏条件下,杂质C 均显著增加,在光破坏条件下产生2 个未知杂质,所产生的降解产物在该色谱条件下能与主峰有效的分离。具体见图2。

2.4 空白辅料干扰试验
各企业的处方组成基本一致,以亚硫酸氢钠或焦亚硫酸钠作为抗氧剂,以依地酸二钠作为金属离子螯合剂,处方量略有不同; 个别企业( C 公司) 添加苯甲醇作为止痛剂。
称取各企业处方中各辅料的最大取样量制成混合空白辅料溶液,分别精密量取混合辅料溶液及空白溶剂各5 μl,依法测定,结果见图3。

结果表明: 空白辅料在保留时间3.7 min前( 相对安乃近保留时间0.25倍处) 存在辅料峰,而C 公司的处方中的特殊辅料( 苯甲醇) 在保留时间约17.353 min( 相对安乃近保留时间1.19倍处) 存在较大的辅料峰; 辅料峰不干扰有关物质的检测。
2.5 校正因子的确定
4-甲氨基安替比林( 杂质C) 及4-N-去甲基安乃近( 杂质E) 是样品中检出的主要已知杂质,对这2 个杂质相对安乃近的校正因子进行了考察。
分别称取安乃近、杂质C 及杂质E 对照品,用无水甲醇溶解并稀释制成浓度约为40,60,80,100,120,140,160 μg·ml-1 的溶液,精密量取5 #l 进样。
由峰面积对溶液浓度作线性回归,得回归方程、相关系数、斜率和截距。用安乃近回归方程的斜率与杂质回归方程斜率的比值计算各杂质的校正因子。
结果见表1。

2.6 定量限及检出限
取安乃近、杂质C 及杂质E 对照品各适量,通过逐级稀释,进样测定,确定安乃近、杂质C 及杂质E 的定量限( S /N≈10) 分别为0.427,0.684,0.372 μg·ml-1 ; 检出限( S /N≈3) 分别为0.113,0.126,0.091 μg·ml-1。
2.7 溶液的稳定性试验
取安乃近、杂质C 及杂质E 对照品适量,分别用无水甲醇溶解并稀释制成50 μg·ml-1 的溶液; 另取供试品( N 公司,批号: 1606067 2) 1.0 ml,用无水甲醇溶解并稀释制成5mg·ml-1 的溶液。
取上述溶液分别于0,1,2,4,6,8h 取样测定。结果显示,安乃近、杂质C 及杂质E 对照品溶液主峰面积RSD 分别为0.10%、1.22% 及0.89% ( n = 6) ,3种溶液8 h 内基本稳定; 供试品溶液中杂质C 的峰面积随时间不断增加,8h 内RSD 为23.6% ( n = 6) ,建议供试品溶液临用新制。
2.8 耐用性试验
取“2.2”项下系统适用性溶液,在保持其他色谱条件不变的情况下,更换仪器( Agilent 1260 和Dionex Ultimate3000) ,色谱柱品牌( Xtimate、ZORBAX 和InertSustain) ,调整柱温( 30,35,40℃) 与流速( 0.9,1.0,1.1 ml·min-1 ) ,以考察测定条件有微小变动时,测定结果不受影响的程度。
结果表明,在改变上述色谱条件的情况下,安乃近的理论塔板数均大于5 000,与相邻杂质峰的分离度均大于1.5,方法耐用性良好。
2.9 样品测定
取本品适量( 约相当于安乃近250 mg) ,置50 ml 量瓶中,加无水甲醇稀释至刻度,摇匀,作为供试品溶液( 临用新制) ;
精密量取1 ml,置100 ml 量瓶中,加无水甲醇稀释至刻度,摇匀,作为对照溶液。精密量取对照溶液5 μl 注入HPLC 仪,调节检测灵敏度,对照溶液的主峰高为满量程的20%;
再精密量取供试品溶液和对照溶液各5 μl,注入HPLC仪,记录色谱图至主成分峰保留时间的3.5 倍,扣除辅料峰,已知杂质采用加校正因子的自身对照法计算,未知杂质采用自身对照法计算。取15 家生产企业各1 ~3 批样品,依法测定,结果见表2。

3
讨论
3.1 检测波长的选择
截取“2.2”项下系统适用性溶液二极管阵列检测( DAD) 图中安乃近与其主要中间体及杂质的紫外光谱图,结果安乃近在263 nm 波长处有最大吸收,主要中间体及杂质的最大吸收波长各不相同,参考EP 8.0[11],选择254 nm作为有关物质检测波长,在该波长下,安乃近与其主要中间体及杂质均有较大的吸收,能有效的检出。
3.2 稀释溶剂及进样量的选择
安乃近易溶于水,但在水中极易水解为杂质C,而在无水甲醇中相对较为稳定,故选择无水甲醇作为溶剂。
由于选择无水甲醇作为溶剂,与流动相系统存在较大差异,存在较强的溶剂效应,参考EP 8.0中安乃近有关物质检查方法[11],将进样量降为5 μl,获得保留时间、理论板数、对称性较好的色谱峰。
3.3 杂质的来源分析
本品共检出2 个已知杂质( 杂质C 及杂质E) 及4 个未知杂质。
杂质E 为原料药合成副产物,每批样品均有检出,杂质量均小于0.1%,符合EP 8.0 该杂质的限度要求( 0.15%) 。
杂质C 既是安乃近合成中间体,也是水解产物,同时也是活性代谢物,每批样品均有检出,杂质量在2.87 ~5.82%之间,参照安乃近原料药现行标准杂质C 限度( 0.5%) 及安乃近片现行标准[10]杂质C 限度( 1.0% ) 拟订本品的杂质C 限度为1.0%,38 批样品均不符合规定。已知杂质C 及E 的结构见图4。

3.4 杂质的毒性分析
通过美国Simulations Plus 公司的ADME/T 性质预测软件ADMET PredictorTM ( Version 8.1.0.11,Simulations Plus,Inc.,USA) ,对安乃近及已知杂质的毒理做全面预测: 三者的整体毒性风险打分均小于3.3( 该风险系数越大,表示化合物的毒性性质越不理想,建议毒性风险≤3.3) ,风险较低; 但三者毒性存在差异,安乃近和杂质E 未表现出明显毒性; 杂质C 表现出小鼠致癌性和一定的致突变性,整体毒性比较杂质C > 杂质E≈安乃近。